Article Text

Abstract
Introduction Idiopathic nephrotic syndrome is the most common glomerular disease in childhood with an incidence of 1.8 cases per 100 000 children in Germany. The treatment of the first episode implies two aspects: induction of remission and sustainment of remission. The recent Kidney Disease Improving Global Outcomes, American Academy of Pediatrics and German guidelines for the initial treatment of the first episode of a nephrotic syndrome recommend a 12-week course of prednisone. Despite being effective, this treatment is associated with pronounced glucocorticoid-associated toxicity due to high-dose prednisone administration over a prolonged period of time. The aim of the INTENT study (Initial treatment of steroid-sensitive idiopathic nephrotic syndrom in children with mycophenolate mofetil versus prednisone: protocol for a randomised, controlled, multicentre trial) is to show that an alternative treatment regimen with mycophenolic acid is not inferior regarding sustainment of remission, but with lower toxicity compared with treatment with glucocorticoids only.
Methods and design The study is designed as an open, randomised, controlled, multicentre trial. 340 children with a first episode of steroid-sensitive nephrotic syndrome and who achieved remission by a standard prednisone regimen will be enrolled in the trial and randomised to one of two treatment arms. The standard care group will be treated with prednisone for a total of 12 weeks; in the experimental group the treatment is switched to mycophenolate mofetil, also for a total of 12 weeks in treatment duration. The primary endpoint is the occurrence of a treated relapse within 24 months after completion of initial treatment.
Ethics and dissemination Ethics approval for this trial was granted by the ethics committee of the Medical Faculty of the University of Heidelberg (AFmu-554/2014). The study results will be published in accordance with the Consolidated Standards of Reporting Trials statement and the Standard Protocol Items: Recommendations for Interventional Trials guidelines. Our findings will be submitted to major international paediatric nephrology and general paediatric conferences and submitted for publication in a peer-reviewed, open-access journal.
Trial registration number DRKS0006547; EudraCT2014-001991-76; Pre-result.
Date of registration 30 October 2014; 24 February 2017.
- mycophenolate mofetil
- steroid-sensitive nephrotic syndrome
- steroids
- alternative treatment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first trial worldwide that prospectively evaluates a steroid-reduced initial treatment alternative for childhood nephrotic syndrome.
This trial has the potential to reduce steroid-associated side effects without losing efficacy.
If our hypotheses turn out to be true, the experimental therapy has the potential to become the future standard of care.
This is one of the few randomised, controlled, prospective, multicentre trials in paediatric nephrology, but due to clinical and financial aspects the trial is not blinded.
Introduction
Idiopathic nephrotic syndrome in childhood
Clinical course and epidemiology
Idiopathic nephrotic syndrome in childhood, defined as the combination of heavy proteinuria (>40 mg/m2 body surface area (BSA) per hour) and hypoalbuminaemia (<25 g/L), in general accompanied by oedema and hyperlipidaemia, is a rare, relapsing disease with an incidence of 1.8 per 100 000 children below 16 years of age in Germany (German registry of rare paediatric diseases, ESPED (Erhebungseinheit für Seltene Pädiatrische Erkrankungen in Deutschland) 2005–2006), resulting in an annual rate of 200–250 new patients.1 The classification according to the four following categories is important for the diagnostics, treatment and prognosis of nephrotic syndrome in childhood: aetiology, age at onset, histology and response to glucocorticoids. The primary idiopathic nephrotic syndrome with a typical onset at 1–10 years of age should be differentiated from patients with secondary causes or patients with age at onset younger than 1 year (congenital and infantile forms) or older than 10 years of age. Approximately 80% of children with idiopathic nephrotic syndrome have minimal change disease on renal biopsy and approximately 7% have focal segmental glomerulosclerosis. The most important prognostic factor is steroid sensitivity occurring in over 90% of the patients.
Treatment
The treatment of the first episode implies two aspects: induction of remission and sustainment of remission. The Gesellschaft für Pädiatrische Nephrologie (GPN), formerly known as Arbeitsgemeinschaft für Pädiatrische Nephrologie, defined the standard of care for children with nephrotic syndrome.2–7 The effectual guideline for the initial treatment of the first episode of a nephrotic syndrome recommends in detail 60 mg prednisone/m2 BSA per day (maximum 80 mg/day) for 6 weeks followed by alternate-day prednisone 40 mg/m2 BSA (maximum 60 mg/48 hours) for another 6 weeks.8
In case of steroid sensitivity remission usually occurs within 7–14 days of treatment; the overall duration of initial prednisone treatment is 12 weeks in order to sustain remission. This regimen is associated with a relapse rate of 51% within 24 months after initial prednisone therapy, and a rate of frequent relapses (definition: relapses occur four or more times in any 12-month period, or two or more relapses within the first 6 months period after initial response) of 29% is expected.
Side effects of treatment
Despite being effective, this treatment is associated with pronounced glucocorticoid-associated toxicity due to high-dose prednisone administration over a prolonged period of time.
The major side effects, which have been shown consistently in previous studies,3 4 9 comprise obesity, striae, hypertrichosis, cataract, glaucoma, arterial hypertension, psychological disturbances, growth failure, disturbances in carbohydrate and lipid metabolism, osteopaenia, and avascular bone necrosis. Not all of these side effects are completely reversible after cessation of steroid therapy. In one study, for example, excessive gain of weight during initial steroid therapy persisted in a significant subset (47%) of patients following cessation of glucocorticoid therapy.10 Obesity following cessation of glucocorticoid therapy was associated with hyperlipidaemia, which might enhance the cardiovascular risk of these patients in the long run.11 Other studies have shown that exposure to higher doses of glucocorticoids in the initial therapy leads to more toxicity without prevention of future relapses.12–15
Role of mycophenolate mofetil in the treatment of nephrotic syndrome in childhood
Mycophenolate mofetil (MMF), the prodrug of its active moiety mycophenolic acid (MPA), is a potent, selective and reversible inhibitor of inosine monophosphate dehydrogenase, the key enzyme of de novo purine synthesis in activated lymphocytes. MMF is effective in sustaining remission in patients with frequently relapsing or glucocorticoid-dependent nephrotic syndrome.
Four prospective studies in patients with frequently relapsing or glucocorticoid-dependent nephrotic syndrome receiving a long-term therapy with MMF explored the possibility of withdrawing prednisone, which was successful in >50% of patients without further relapses.16–19
In children with glucocorticoid-dependent nephrotic syndrome on MMF, Dorresteijn et al 20 reported relapse rates of 25% after 6 months and 45% after 12 months, respectively. In a phase II Bayesian trial, Baudouin et al 21 confirmed the effect of MMF in reducing relapse rates and in sparing glucocorticoids in children with glucocorticoid-dependent nephrotic syndrome. A recent GPN study on the maintenance of remission in children with frequently relapsing or steroid-dependent nephrotic syndrome has shown that MMF in adequate exposure is as effective as ciclosporin A (CSA) in sustaining remission without the burden of CSA-induced nephrotoxicity.22
So far, no studies with MMF for the initial treatment of the steroid-sensitive nephrotic syndrome (SSNS) in children have been performed. However, it seems coherent to use the efficacy of MMF also for sustaining remission in the initial treatment of SSNS and to benefit from its lower toxicity compared with glucocorticoids.
Rationale
The initial treatment of the idiopathic nephrotic syndrome in children requires sufficient immunosuppressive therapy, but should avoid toxicity, since the intensity of the initial treatment does not influence the long-term course of the disease. For example, a GPN trial on the initial treatment of nephrotic syndrome revealed no overall advantage of an intensified immunosuppressive protocol adding CSA in terms of occurrence of relapses during a follow-up of 24 months.5 12 13
Our hypothesised novel treatment protocol has the potential to reduce the burden of glucocorticoid-associated side effects and associated cardiovascular risk factors, if the novel protocol is not inferior to the standard therapy regarding sustainment of remission. If our hypotheses turn out to be true, this novel therapy has the potential to become the standard of care for the initial treatment of SSNS in children.
Methods/Design
Aim
The main purpose of the study is to show that MMF in the initial treatment of SSNS in children is not inferior regarding maintenance of initial remission and subsequent relapse rate compared with the standard prednisone regimen.
Study design
This is a prospective, randomised, multicentre, controlled, open, parallel-group, phase III, non-inferiority trial.
After initiation of the study, patients will be screened consecutively and eligible patients will be enrolled into the study at each centre.
Each site’s principal investigator has to declare to the coordinating investigator/sponsor that he/she will conduct the study according to the protocol and ethical rules, and to provide the support as needed. To minimise a potential performance bias, this will be fixed in a contract prior to commencing the study. The clinical monitor will introduce the sites in detail to study procedures and documentation in advance.
Bias by potential influential factors will be addressed by inclusion as covariates into the statistical analysis. Independent clinical onsite monitoring to ensure patients’ safety and integrity of the clinical data in adherence to study protocol will focus on source data documentation, correctness of data and adherence to study procedures, for example, randomisation and treatment.
Based on the performed interventions and planned analysis, blinding is not feasible to minimise bias, because the interventions can easily be differentiated due to visible side effects such as obesity, which is only expected in the standard care group. Furthermore, MMF is used in liquid form as a suspension and prednisone as a tablet. However, the primary endpoint is based on standardised diagnostic work-up results, that is, objective criteria.
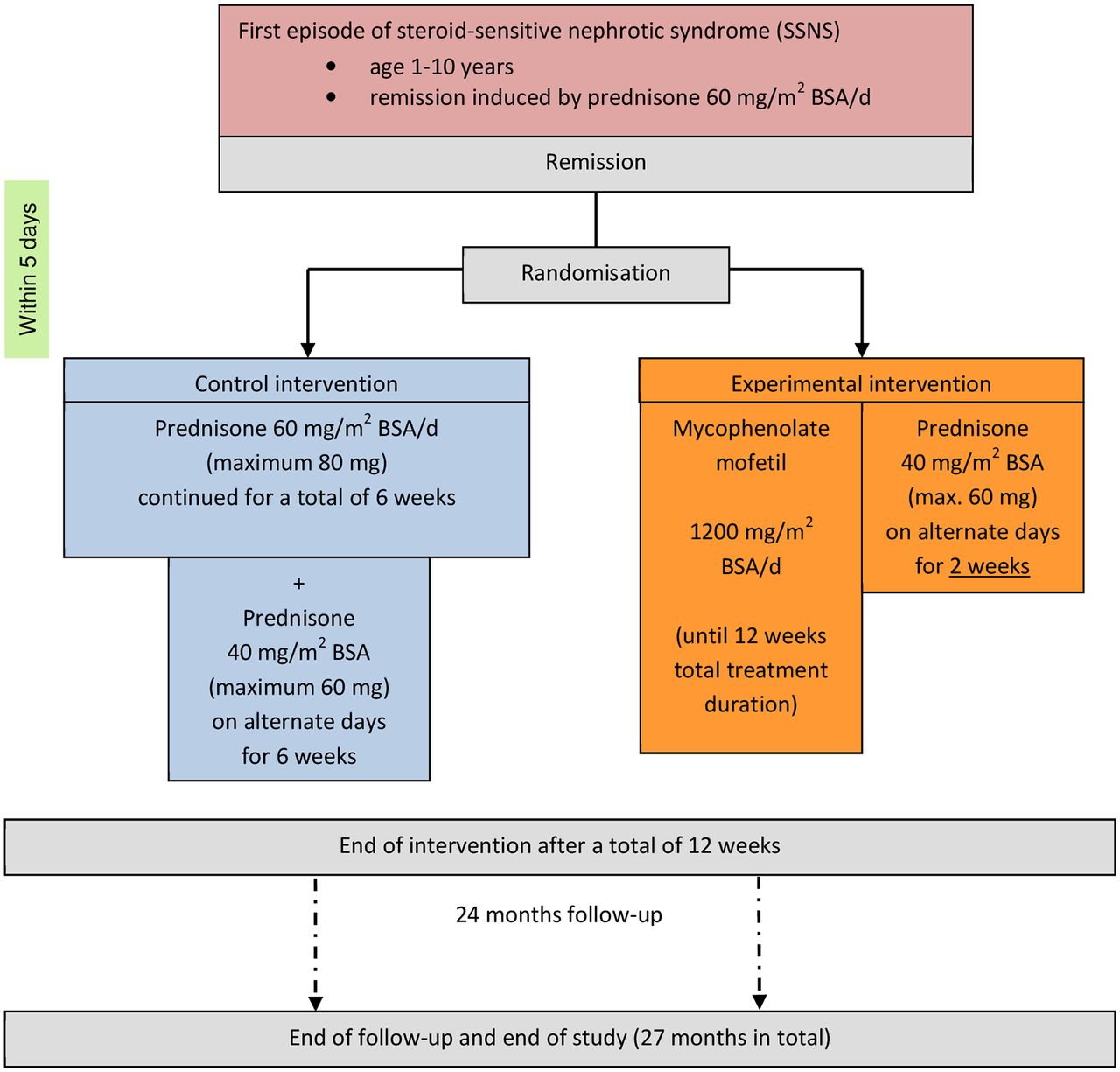
The duration of the study for each subject is expected to be 27 months (including 24-month follow-up after intervention) (figure 1 and figure 2).

Trial schema. On alternate days means every second day. BSA, body surface area.

{kind=link}
{kind=link}
Study visit schedule. *Only in the experimental group. ABPM, ambulatory blood pressure monitoring; HRQoL, health-related quality of life; TDM MPA, therapeutic drug monitoring of mycophenolic acid.
Patient and public involvement
Patients were not directly involved in the study development and design. Repeated discussions with patient representatives beforehand showed one of their main wishes is reduction of steroids in the treatment of nephrotic syndrome.
We generated an information document for parents in the form of a flyer which was also distributed to patient initiatives. Spreading out information on the study shall improve recruitment. There is no patient adviser involved in the conduct of the study, neither was the burden of the intervention assessed by patients or their parents during study development.
Study results will be published open access. Patients and their representatives will be informed through meetings and a brief summary of the results distributed by local investigators.
Recruitment
The study is conducted on a multicentre basis. The rarity of the disease requires a nationwide recruitment. The planned 35 study centres are evenly distributed over Germany. Each study centre is coordinating a number of collaborating hospitals and practitioners that will transfer eligible patients with primary onset SSNS for screening, enrolment, randomisation and study visits. Four hundred patients should be assessed for eligibility, and 340 subjects should be enrolled in the clinical study, that is, 170 subjects per treatment group.
Inclusion criteria and exclusion criteria
Inclusion criteria
Subjects meeting all of the following criteria will be considered for admission to the study:
First episode of SSNS.
Remission induced by prednisone or prednisolone 60 mg/m2 BSA (maximum 80 mg/day) per day within 28 days.
Male and female children aged ≥1 year and ≤10 years at beginning of the study (typical age range of patients with SSNS).
Ability of the persons having care and custody of the child to understand character and individual consequences of clinical study.
Written informed consent of the persons having care and custody of the child (must be available before enrolment in the study).
Exclusion criteria
Subjects presenting with any of the following criteria will not be included in the study:
Secondary nephrotic syndrome.
Estimated glomerular filtration rate (eGFR) <90 mL/min×1.73 m2 BSA.
Ongoing treatment with systematically administered glucocorticoids or other immunosuppressive drugs at the time of the first episode of nephrotic syndrome.
Haemoglobin concentration of ≤90 g/L (SI unit).
Leucocyte count of ≤2.5×109/L (SI unit).
Severe chronic gastrointestinal disease.
History of hypersensitivity to MMF or to any drug with similar chemical structure or to any excipient present in the pharmaceutical form of suspension of MMF (CellCept suspension).
Refusal of subject.
Participation in other clinical studies or observation period of competing studies.
Study medication
The sponsor, that is, the University Hospital Heidelberg, will provide the required study medication (MMF, CellCept suspension). Careful records will be kept of the study medication supplied to the centres and distributed to the patients.
Prednisone is used as standard therapy following the definition of the GPN (standard treatment) and is prescribed as usual.
Prednisone or prednisolone (control intervention).
MMF is administered in liquid form (CellCept suspension (Roche Registration)) (experimental intervention).
Adherence
Adherence will be recorded by patients’ diary.
Screening
All patients who seem suitable for study participation and take part in the screening will receive a screening number and will be registered in a screening log. Together with the centre ID, this will be the unique identification number throughout the study.
Parents of children with initial episode of idiopathic nephrotic syndrome aged between 1 and 10 years and treated with standard regimen (prednisone 60 mg/m2 BSA per day) will be informed about the ongoing INTENT study. If the child fulfils the inclusion criteria, the persons having care and custody of the child and the patient, if ≥6 years of age, will be formally elucidated about the INTENT study by the study centre in a form understandable to him or her and asked for written assent/consent.
For checking the exclusion criteria concerning eGFR, leucocyte count and haemoglobin concentration, the most recent lab values should be used; they should have been obtained no more than 28 days prior to visit 1.
Randomisation
To achieve comparable intervention groups, patients will be allocated in a concealed fashion by means of randomisation using a centralised web-based tool (www.randomizer.at). Randomisation will be performed stratified by age groups (grouped: <7 years of age, ≥7 years of age), because age is known to influence the occurrence of relapses. If the randomiser is not available in urgent cases, the Institute of Medical Biometry and Informatics (IMBI) can be contacted and a biometrician or data manager will perform the randomisation.
Intervention
The maximum duration of treatment is 12 weeks after the first day of initial treatment of SSNS (figure 1).
Control intervention
Prednisone, which is continued for a total of 6 weeks with a dosage of 60 mg/m2
BSA/day (maximum 80 mg), is given twice per day or three times per day.
plus
Prednisone, which is given for another total of 6 weeks with a dosage of 40 mg/m2
BSA (maximum 60 mg) on alternate days (every other day) in one dose in the morning.
Resorption of prednisone is independent of food intake.
Experimental intervention
MMF is given in a dosage of 1200 mg/m2 BSA/day as a suspension (200 mg/mL) until 12 weeks of the total treatment duration. MMF is given twice a day, that is, every 12 hours (±1 hour).
The suspension of MMF is prepared in the study centre (according to the summary of product information).
The persons having care and custody of the child are informed that MMF should be given 30 min before or 60 min after food intake.
For the first 2 weeks from randomisation, prednisone is given with a dosage of 40 mg/m2 BSA (maximum 60 mg) on alternate days (every other day) in one dose in the morning.
At visits 2 and 3 MPA exposure is measured by a limited sampling strategy (blood samples are obtained at time points 0, 1 and 2 hours after intake of MMF).
Recording of primary endpoint
Daily dipstick testing of urine (Albustix) and documentation in a standardised diary by a person having care and custody of the child are a common current practice in the care of patients with nephrotic syndrome in paediatric nephrology centres.
No guideline exists on whether standard relapse treatment with prednisone should be started immediately when the definition of relapse is fulfilled to avoid the associated complications of an oedematous relapse or whether treatment should be delayed for several days to determine whether proteinuria resolves spontaneously. Therefore, in the INTENT study a time period of up to 10 days is allowed for a possible spontaneous remission, before standard therapy for relapse is started.
Treatment of a relapse has to be performed according to standard therapy of the GPN (prednisone 60 mg/m² BSA (maximum 80 mg) per day until the urine is free of protein for three consecutive days, followed by alternate-day prednisone 40 mg/m² BSA (maximum 60 mg) for 4 weeks). Relapses with and without treatment are documented on the electronic case report form (eCRF).
Treatment of frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome with other medications than prednisone is carried out according to centre practice, because there is no internationally accepted guideline on this topic. The performed treatment with immunosuppressive agents such as CSA, tacrolimus, MMF, cyclophosphamide, rituximab or levamisole is documented on the eCRF.
After completion of the study, patients will be treated according to centre practice.
Outcome measures
Primary study endpoint
The primary efficacy endpoint is occurrence of a treated relapse within 24 months after completion of initial treatment. The rationale is that this endpoint was chosen in all previous studies on the initial treatment of SSNS in children and is also the primary endpoint in various meta-analyses on this topic.3–5 7 8
Definition of relapse: Relapse is denoted by a reappearance of proteinuria for three consecutive days:
Albustix ≥2+ (first or second morning urine).
or
Urine protein:creatinine ratio ≥2 g/g (first or second morning urine).
or
Urine protein excretion of ≥40 mg/m2 BSA/hour (urine collection for a minimum of 12 hours).
Relapses with and without treatment are documented. The primary endpoint is fulfilled by the first treated relapse.
Secondary endpoints
Secondary endpoints are divided into five items:
Course of the disease as described by the following criteria:
Time from remission to first relapse.
Number of relapses during follow-up.
Mean relapse rate per patient and year.
Number of frequent relapsers.
Time from remission to intensification of immunosuppressive treatment with other drugs due to glucocorticoid-induced toxicity.
Rate of patients who require more intense immunosuppressive treatment (eg, CSA, tacrolimus, MMF, cyclophosphamide, rituximab or levamisole).
Glucocorticoid-associated toxicity:
Cumulative prednisone dose as mg/m2.
As there is no validated score for glucocorticoid-induced toxicity, each item is registered separately. At study visits 1–8, body mass index, blood pressure and growth will be checked for quantitative influence, striae, hypertrichosis, acne and psychological disturbances by yes or no for qualitative influence. Additionally, at study visits 1, 5 and 8, patients will be checked for cataract and glaucoma (by yes or no).
MMF-associated toxicity: At all visits, patients will be checked for known side effects of MMF, especially diarrhoea, blood cell count disturbance and infections.
Health-related quality of life, which may be impaired in children with nephrotic syndrome, will be measured with a validated questionnaire (DISABKIDS) at visits 1/5/8.
Days missing school attendance and days of hospitalisation will be documented as a measure of the impact of the disease on everyday life.
It is expected that the MMF-based regimen will avoid acute and long-term glucocorticoid-associated toxicity and is therefore superior regarding the benefit:risk ratio. However, this will not be tested confirmatorily, since there is no endpoint or score summarising the different aspects of side effects.
Statistical considerations
Sample size calculation
The sample size calculation is based on the primary efficacy endpoint ‘occurrence of a treated relapse within 24 months after completion of the initial treatment’. In the literature varying information is given regarding the relapse rate for the control group receiving standard prednisone therapy. We have decided to assume a relapse rate of 51% according to Gipson et al.8 The same rate is expected for the experimental group. If the relapse rate in the experimental group accounts to less than 15% above the relapse rate of the control group, this will be considered as clinically irrelevant based on clinical judgement. Therefore the margin is set to δ=0.15. As the direction of the difference to be established is known for non-inferiority studies and as—due to the rareness of the disease and the related limited available number of patients—the study could otherwise not be performed with sufficient power, a one-sided significance level of 5% is applied. Testing at a one-sided significance level of α=5% and aspiring a power of 80%, a total of 272 patients (136 per group) are required (calculations performed with ADDPLAN V.6.0). To account for a 10% dropout rate and major protocol violations in a further 10%, 340 patients will be randomised.
Adherence/rate of loss of follow-up
The nephrotic syndrome in children is mostly an acutely presenting disease, and parents are very concerned about their child. With standard prednisone treatment, we observe a high adherence to therapy. According to our previous experience in performing studies in paediatric nephrology, we assume that a minimum of 85% of patients assessed for eligibility will be allocated to the study.4 5 22 Due to the exclusive care of these patients in specialised paediatric nephrology centres, we calculate a loss of follow-up either due to dropout or major protocol violation of a maximum of 20%, which corresponds to our previous studies.4 5 22 The recent study of the GPN, showing that MMF is efficacious in sustaining remission in children with frequently relapsing nephrotic syndrome, had only a dropout rate of 4%. Therefore, for the entire study, we estimated 400 children with SSNS to be assessed for eligibility, 340 to be allocated to the study and 272 patients to be analysed per protocol. However, in cases of premature withdrawal by a patient, the persons having care and custody of this patient will be asked for informed consent so that routinely recorded data by the covering physician can be used for the INTENT study. In this manner as many data as possible are recorded for evaluation of treatments in this rare disease.
Analysis populations
The primary analysis will be performed for both the per-protocol (PP) population and the intention-to-treat (ITT) population. The PP population comprises all patients who were treated according to the randomised treatment as outlined in the protocol without major protocol violations (eg, reduction of study medication of >50% or interruption of study medication of >3 days, violation of inclusion or exclusion criteria). The ITT population will comprise all patients randomised into the study. In this set, every patient is analysed according to the group randomised into.
Since there may be patients who withdraw from the study after the treatment period or within the treatment period but consent to the analysis of routinely recorded data was given, the inclusion of these patients into the ITT population will be decided case by case before database lock and defined when writing the statistical analysis plan (SAP). As appropriate, a third population will be defined for analysis of the primary and important secondary endpoints. How to deal with these patients and their data in detail depends on the time point of withdrawal and the amount and reliability of the routinely collected data.
The safety set will comprise all patients who have received study medication at least once, and will allocate the patients to the treatment they actually received, regardless of randomisation. Whether routinely collected data of patients who withdraw prematurely can be included herein depends on the reliability of the collected safety data.
Statistical methods
The non-inferiority of the experimental group versus the control group will be evaluated using the test according to Farrington and Manning. The one-sided significance level is set to 5%.
The hypotheses to be assessed in the primary efficacy analysis are formulated as follows:
H0: p_MMF – p_Prednisone ≥ δ (δ=0.15, non-inferiority margin, see sample size calculation for justification).
H1: p_MMF – p_Prednisone < δ, where p_* denotes the relapse rate in the respective group.
Before database closure the assignment of patients to the PP population (patients with no major protocol violations) and the ITT population (as classified by the ITT principle) is defined in the SAP. The confirmatory analysis is performed for both the PP population and the ITT population. This approach reflects the equal importance of both analysis sets in a non-inferiority trial. For the PP analysis missing values for the primary endpoint are not expected. In the ITT population missing values will be replaced according to Higgins et al.23 As appropriate, a third population will be defined to adequately incorporate routinely collected data of patients who withdraw prematurely but gave informed consent for usage of routinely collected data. Details on inclusion of such data into sensitivity analyses of primary and secondary endpoints will be defined in more detail in the SAP. In case of uncertainty regarding data quality and reliability, these patients will only be analysed descriptively.
Additionally, binary logistic regression models will be performed as sensitivity analysis for the intervention comparison of the relapse rates adjusting for age, gender, centre (grouped), and for results of therapeutic drug monitoring (grouped) based on different populations (PP and ITT, with values of dropouts set to worst case).
All secondary outcomes will be evaluated descriptively, using appropriate statistical methods based on the underlying distribution of the data. Descriptive p values are reported together with 95% CIs for the corresponding effects. Descriptive statistics for continuous parameters and scores include the number of non-missing observations, mean, SD, median, minimum and maximum, performed for treatment groups as well as subgroups and overall. The description of categorical variables (ordinal or nominal) includes the number and percentage of patients belonging to the relevant categories in the study population as well as to each treatment group.
Rates of adverse events (AEs) and serious adverse events (SAEs) will be calculated with 95% CI for treatment group comparisons.
Statistical methods are used to assess the quality of the data, homogeneity of treatment groups, endpoints and safety of the two intervention groups. Details of the statistical analysis will be fixed at the latest in the SAP to be prepared within the first year after the start of patient recruitment. All persons taking part in the preparation of the SAP and possible later changes to it will only have access to blinded data to avoid introduction of bias.
Interim analyses
No interim analysis will be performed for the following reason: The recruitment phase is planned to be 36 months. The primary endpoint is occurrence of treated relapse within 24 months after end of initial treatment. Therefore, information on the primary endpoint for a first portion of the study patients will be available not before the end of the recruitment phase. For this reason, a group-sequential approach was not pursued.
However, an independent data safety monitoring board will closely monitor the recruitment, the reported AEs, the data quality of the study and the occurrence of potential early relapses during the intake of MMF, thus ensuring the ethical conduct of the study and protecting the safety interests of patients.
Adverse events
AEs will be ascertained by the investigators using non-leading questions, noted as spontaneously reported by the patients to the medical staff or observed during any measurements on all study days. The observation period begins with the first administration of the investigational medicinal product and ends with visit 4 (ie, 6 months after day 1 (=first day of treatment with standard therapy)). The patient or his primary care physician should report any AE during the outpatient period via phone to the investigator.
AEs will be documented on the patient file and on the eCRF. All subjects who present AEs, whether considered associated with the use of the study medication or not, will be monitored by the responsible investigator to determine their outcome; this applies to withdrawals, too.
All SAEs and their relevance for the benefit–risk assessment of the study will be evaluated continuously during the study and for the final report.
All SAEs must be reported by the investigator to the Department of Pharmacovigilance at the Coordination Centre for Clinical Trials (KKS) Heidelberg within 24 hours after the SAE becomes known using the ‘Serious Adverse Event’ form.
Suspected unexpected serious adverse events are to be reported to the responsible ethics committee, the competent authority and to all participating investigators within defined timelines, that is, they are subject to an expedited reporting.
All SAEs will be subject to a second assessment by a designated person or his deputy, who will be independent from the reporting investigator.
Data management
Data management and quality assurance
The investigator or a designated representative must enter all protocol-required information in the eCRF. The eCRF should be completed as soon as possible after the information is collected, preferably on the same day when a study subject is seen for an examination, treatment or any other study procedure. The reason for missing data should be provided. The investigator is responsible for ensuring that all sections of the eCRF are completed correctly and that entries can be verified in accordance with the source data. Any entry and correction in the Remote Data Entry System will be documented automatically in an audit file.
Completeness, validity and plausibility of data will be checked in time of data entry (edit-checks) and using validating programs, which will generate queries. The investigator or the designated representatives are obliged to clarify or explain the queries. If no further corrections are to be made in the database, it will be closed and used for statistical analysis. All data management procedures will be carried out on validated systems and according to the current standard operating procedures of the IMBI of the University of Heidelberg.
Ethical and legal aspects
The procedures set out in this study protocol, pertaining to the conduct, evaluation and documentation of this study, are designed to ensure that all persons involved in the study abide by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use harmonised tripartite guideline on Good Clinical Practice (ICH-GCP) and the ethical principles described in the applicable version of the Declaration of Helsinki.
The study will be carried out in conformity with the ICH Topic E6, Guideline for Good Clinical Practice, including post step 4 errata, September 1997, Directive 2001/20/EC (4 April 2001), Commission Directive 2005/28/EC (8 April 2005), national regulatory requirements/guidelines of the participating countries concerning clinical studies (eg, federal drug law (AMG (Arzneimittelgesetz, German Medicinal Products Act), GCP ordinance (GCP-Verordnung), medical device law (MPG (Medizinproduktegesetz, Act on Medical Devices)), and general national regulatory requirements, for example, Bundesdatenschutzgesetz (BDSG).
Approval of the regulatory authorities
According to the German Federal law the study was approved by the Federal Institute of Drugs and Medical Devices on 2 April 2015 (reference number 61-3910-4040246). The latest version of the trial protocol (V.5.0) was approved by the Federal Institute of Drugs and Medical Devices on 11 July 2016.
Discussion
Risk–benefit assessment
Neither intensification nor prolonging initial therapy has influenced long-term prognosis of SSNS in terms of the number of relapses and risk of frequent relapses.12–15 MMF is effective in sustaining remission in patients with frequently relapsing SSNS.16 21 22 Therefore we hypothesise that after initial remission is achieved, the risk for immediate relapse will not be increased in the experimental group. If a patient of the experimental group develops a relapse under MMF therapy, he or she will be given prednisone anyway for induction of remission; the overall prognosis would therefore not be influenced. On the other hand, the patients in the experimental group may have the potential to benefit significantly because of less glucocorticoid-associated toxicity.
The most frequently observed side effects of MMF are gastrointestinal symptoms such as nausea, vomiting, stomach pain and diarrhoea, and haematological symptoms such as leucopenia, anaemia and rarely thrombocytopaenia and an enhanced susceptibility for infections. In general, these side effects occur more frequently and have a higher clinical significance, when MMF is administered in conjunction with other immunosuppressive medication such as CSA or tacrolimus, as indicated after solid organ transplantation.
When MMF is administered as monotherapy, for example, in patients with frequently relapsing SSNS, the frequency and severity of these side effects are markedly lower.16–21 Side effects will be systematically evaluated during the trial visits.
In order to acknowledge recently reported AEs (hypogammaglobulinaemia, bronchiectasis, the risk of teratogenicity and mutagenicity) in patients after solid organ transplantation and treated with MMF in conjunction with other immunosuppressive medications in the long-time run, these AEs are also monitored closely in the INTENT study, even though these events are very unlikely to occur due to the short administration period of MMF (maximum 11 weeks) and the age group being tested in this trial.
The oral formulation of MMF being a suspension allows exact and flexible dosing and reliable administration even to small children.
Cost–benefit analysis
The costs for a treatment with MPA for an average time of 74 days (84 days of initial treatment minus an average of 10 days until remission) in a child with a BSA of 0.8 m² in Germany are approximately ten times higher than the standard treatment with prednisone (€500 compared with €50). With the expected 250 new cases of childhood nephrotic syndrome per year, this would mean extra costs of about €110 000 for the German healthcare system. On the other hand, it has been shown that excessive weight gain during the initial steroid therapy in a significant subset (47%) of patients after cessation of glucocorticoid therapy persisted and thus contributes to long-term cardiovascular risk.10 11 These potential extra costs are hardly to be calculated, but it seems reasonable enough to avoid long-term effects of high-dose prednisone treatment.
Potential impact
The current study continues the long-lasting tradition of prospective randomised trials on the initial treatment of idiopathic nephrotic syndrome performed by the GPN (formerly Arbeitsgemeinschaft für Pädiatrische Nephrologie).
This is the first trial worldwide that prospectively evaluates a steroid-reduced initial treatment alternative that has the potential to reduce the number of side effects without lacking efficacy. If our hypotheses turn out to be true, the experimental therapy has the potential to become the future standard of care.
Optimising recruitment
Our structure of numerous study centres covering entire Germany that collaborate with regional hospitals and practitioners should make most new manifestations of idiopathic nephrotic syndrome available to study evaluation.
Nevertheless patient recruitment currently stays behind schedule. One aspect to improve recruitment is initiation of further study centres especially in densely populated areas in Southern Germany. Other aspects are strengthening the motivation of collaborating partners to transfer patients, advertising the study in widely distributed journals, by personal contact via mail and phone, and to introduce the study at all suitable annual conferences. If patient recruitment cannot be increased sufficiently by these measures, the recruitment period has to be prolonged.
Dissemination
The study results will be published in accordance with the Consolidated Standards of Reporting Trials statement and the Standard Protocol Items: Recommendations for Interventional Trials guidelines. Our findings will be submitted to major international paediatric nephrology and general paediatric conferences and submitted for publication in a high-impact factor journal with open access.
Trial status
The recruitment of the study started in October 2015.
As of 12 June 2018 a total of 156 children have been recruited into the study.
References
Footnotes
BT and LTW contributed equally.
RE and MRB contributed equally.
Contributors MRB, LTW, BT, JD, JG, DH, BH, PFH, MJK, MK, UQ, AF, AS and RE designed the study. AS, MRB, RE, BT and LTW will undertake data analyses. BK and SL gave advice on regulatory affairs and on the realisation of the study. RE, MRB, BT and LTW wrote the first draft of this manuscript, which has been critically revised by all coauthors. All authors have read and approved the final version of the manuscript.
Funding The INTENT study is funded by the German Federal Ministry of Education and Research (BMBF, grant 01KG1301).
Competing interests RE, MRB, JD, AF, JG, DH, BH, PFH, BK, MJK, MK, SL, UQ and AS declare to have no competing interests. BT and LTW have received research grants from Roche Pharma AG and Novartis AG.
Patient consent Not required.
Ethics approval Ethics approval of the INTENT study was granted by the ethics committee of the Medical Faculty of the University of Heidelberg (AFmu-554/2014) on 18 March 2015. This approval has subsequently been confirmed by the local ethics committee of all participating centres. The latest version of the trial protocol (V.5.0) was approved by the ethics committee on 1 June 2016. Informed consent will be/has been obtained from all participants.
Provenance and peer review Not commissioned; externally peer reviewed.