Article Text

Abstract
Objectives To characterise postmarketing studies for drugs that were newly approved by the US Food and Drug Administration and the European Medicines Agency.
Design and setting Cross-sectional analysis of postmarketing studies registered in ClinicalTrials.gov until September 2014 for all novel drugs approved by both regulators between 2005 and 2010. Regulatory documents from both agencies were used.
Primary and secondary outcome measures All identified postmarketing studies were classified according to planned enrolment, funding, status and geographical location, and we determined whether studies studied the originally approved indication.
Results Overall, 69 novel drugs approved between 2005 and 2010 were eligible for inclusion. A total of 6679 relevant postmarketing studies were identified; 5972 were interventional (89.4%). The median number of studies per drug was 55 (IQR 33–119) and median number of patients to be enrolled per study was 60 (IQR 28–183). Industry was the primary sponsor of 2713 studies (40.6%) and was a primary or secondary sponsor in 4176 studies (62.5%). In all, 2901 studies (43.4%) were completed, 487 (7.3%) terminated, 1013 (15.2%) active yet not recruiting, 1895 (28.4%) recruiting and 319 (4.8%) not yet recruiting. A total of 80% of studies were conducted in only one country and 84.4% took place in Europe and/or North America; 2441 (36.5%) studied another indication than the originally approved indication. Studies designed in the originally approved indication were found to be more industry-sponsored than others 68.7%vs53.7%; P<0.0001.
Conclusions Postmarketing pharmaceutical research was highly variable and predominantly located in North America and Europe. Postmarketing studies were frequently designed to study indications other than the originally approved one. Although some findings were reassuring, others question the lack of coordination of postmarketing research.
- therapeutics
- epidemiology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first study to systematically assess clinical studies performed after marketing approval by the two leading regulators, namely the US Food and Drug Administration and the European Medicines Agency.
This approach allowed us to examine a substantial number of postmarketing studies over a long time period.
However and due to registration bias, we cannot exclude that some true postmarketing studies were missed and therefore unanalysed.
Introduction
The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) are the two largest and most influential drug regulators worldwide. They tend to maintain similar premarket regulatory standards, and drug manufacturers probably tend to submit the same evidence to both as part of the premarket application process, even though we lack comparative data. Drug evaluation continues after regulatory approval, in particular through postauthorisation requirements and commitments. The US FDA can use several regulatory instruments and harness various sources for postmarketing evaluation of approved drugs. Among them are the FDA Adverse Reporting System and the Sentinel System.1 The EMA also has a set of postauthorisation measures, from direct request by its dedicated committee to specific obligations for certain drugs, all aiming at retrieving data for postmarketing assessment.2 Yet these postmarketing clinical studies required by regulators are limited in number and are not consistently completed.3–5 This situation raises the question of whether other studies of these drugs after regulatory approval, including those conducted by industry and independent investigators, but not to fulfil regulatory requirements, should be considered part of ongoing, continuous evaluation efforts.
Postmarketing studies are designed with different intent than are premarket trials. Their designs are not systematically submitted to regulatory agencies before initiation because many postmarketing studies are conducted by independent investigators, and their conduct is less rigorously regulated.6 Postmarketing studies seek to evaluate safety regarding rare events, to assess the real-life effectiveness of novel drugs and to measure their long-term effects. They also permit drug evaluation in different populations, other indications for the same disease, other diseases or with different delivery systems or dosage forms. Moreover, although premarket trials are nearly exclusively sponsored by the manufacturers, postmarketing studies can be funded by manufacturers but also academic or other types of non-profit institutions. Some research also suggested that a substantial proportion of postmarketing trials, even those with results eventually published in high-impact factor journals, were designed for marketing purposes rather than medical interest.7 8
Nevertheless, postmarketing studies have considerable influence on all stakeholders, in particular researchers, practitioners and regulators or decision-makers, because they provide cumulative evidence regarding marketed products. However, we lack an overall assessment of postmarketing studies regarding novel drugs. Postmarketing research has been studied for high-risk devices9 or even for drugs, but with a focused approach: safety10 11 or given therapeutic areas.12–15 Some of those studies produced reassuring results, yet others showed inconsistencies, with gaps in knowledge regarding some issues.
Our research objective was to provide a comprehensive description of postmarketing studies registered in ClinicalTrials.gov, a publicly accessible clinical trial registry maintained by the US National Institutes of Health over almost a decade for a sample of drugs approved by both the FDA and EMA from 2005 to 2010. We aimed to characterise the total number of studies and patients studied, targeted indications, funding origin, geographical location of studies and status (eg, completed or ongoing). We also sought to examine differences between the condition of the initial label and the specific clinical condition studied in the postmarketing studies, to assess the influence of the sponsor on the targeted indication, and to describe supplemental indications.
Methods
Data sources and study sample
We identified all novel drugs approved between 1 January 2005 and 31 December 2010 by both the FDA and EMA through its Centralised Authorisation Procedure. For the FDA, Drugs@FDA is a publicly accessible database listing relevant regulatory actions for all approved drugs.16 For the EMA, information was accessible in the European Public Assessment Reports, which provide a summary of scientific review and list notable regulatory events for all drug submissions.17 Generic drugs, reformulations, combination therapies and non-therapeutic agents such as radiographic dye were not included. This first search led to a sample of 71 novel drugs approved by both regulators between 2005 and 2010. Two drugs, everolimus and temsirolimus, were excluded because they were associated with an abnormally high number of postmarketing studies involving drug-eluting stents.
Drug and manufacturer characteristics
The following data were retrieved for each drug: agent type (small molecule or biological), dates of regulatory submissions for both the FDA and EMA, orphan status according to the FDA, orphan designation from the EMA, therapeutic class according to the Anatomical Therapeutic Chemical classification,18 initial label from both regulators, degree of novelty (first-in-class, advance-in-class, addition-to-class) as previously described in a paper from FDA officials19 and size of the marketing-authorisation holder (ie, manufacturer). This latter information was obtained by personal communication with EMA officials (Constantinos Ziogas, Small and Medium-sized Manufacturer Office, EMA), who classified manufacturers as large pharmaceutical companies, intermediated-size companies or small and medium-size companies according to the European Union definition based on headcount and financial turnover or balance sheet total.
Preapproval FDA pivotal trial characteristics
We obtained data for the expected length of treatment and number of patients from pivotal efficacy trials supporting FDA approvals that had been collected for a previous work.20 In brief, acute treatment was defined as expected use <1 month, intermediate treatment as expected use from 1 month to 2 years, and chronic treatment as expected use >2 years.
Postmarketing studies
On 24 September 2014, we extracted all studies that were registered at ClinicalTrials.gov for each drug of our sample, regardless of dates and other details. We then excluded studies with the following characteristics: included in the FDA regulatory submission (by a manual review of Drugs@FDA), with inadequate registered status (expanded-access studies, withdrawn studies, suspended studies), and mistakenly extracted (ie, studies actually not assessing the drug of interest). For our main analysis, we decided that all studies whose starting date had preceded the first regulatory submission (to the FDA or EMA) by 1 year or less would be classified as postmarketing studies. However, we also performed most calculations with a slightly different set of studies, namely only those whose launch started after the first regulatory approval of any agency. Trials that pertained to more than one drug in our sample were manually reviewed so as to assign them to only one drug for the sake of further statistical analysis. Clinical judgement was applied to choose the ‘leading’ drug in each study. When we could not determine the leading drug, we used the following rules. If the study was funded by a marketing-authorisation holder of one of the drugs, this drug was considered the leading drug. Otherwise, if the study involved a drug that was assessed for another indication than the originally approved indication, this drug was considered the leading drug. Finally, when no leading drug could be determined, the drug for which the last regulatory approval had been granted was considered the drug tested and was classified as the leading drug.
For all remaining postmarketing studies, the following data were collected: condition studied, starting date, study sponsors (as a primary sponsor or a collaborator), status at the date of extraction (not yet recruiting, recruiting, active yet not recruiting, enrolling by invitation, completed, terminated), number and list of countries, number of centres, study phase, study type (observational or interventional), randomisation and planned enrolment. In addition, studies were classified as assessing the drug for its originally approved indication or not, depending on the initial label. When the initial label differed between the FDA and EMA, we accepted both labels as defining the originally approved indication. One of us (J-DZ) performed this classification after careful review of each primary label. Indications were classified according to the Global Burden of Diseases classification.21 Details of the classification of postmarketing studies are provided in the online supplementary appendix.
Supplementary file 1
Supplemental indications
We also collected approvals of supplemental indications by the FDA during the study period (2005–2014) by manual review of Drugs@FDA. In the ‘Approval date(s) and History, Letters, Labels, Reviews’ section, all events designated as ‘efficacy-new indication’ or ‘efficacy’ were reviewed and retained if deemed appropriate. Labelling revision (such as those related to a modified indication or an expanded patient population) and manufacturing change or addition were not included, nor were irrelevant supplemental indications. We also aimed to assess the average number of patients to be enrolled in postmarketing studies to gain approval of a supplemental indication. For this purpose, we took into account all patients from all postmarketing studies from the start of our sample through 1 year before the issuance of the supplemental indication by the FDA.
Statistical analysis
Using descriptive statistics, we characterised the premarket characteristics of the novel drugs included in our sample (drugs approved by both the FDA and EMA between 2005 and 2010). Next, we used descriptive statistics to characterise features of all identified postmarketing studies registered at ClinicalTrials.gov for all novel drugs. We used a series of trend charts representing the annual number of postmarketing studies over the life-cycle of the drugs according to off-condition and on-condition studies. All statistical tests were two-tailed, with a type I error rate of 0.05. We used SAS V.9.4 (SAS Institute; Cary, North Carolina, USA) for all statistical analyses.
Results
Drug sample
Our study sample included 69 novel drugs approved between 2005 and 2010 by both the FDA and EMA. In all, 51 drugs (73.9%) were small molecules and 18 (26.1%) were biologicals (table 1). The FDA had granted orphan status to 18 drugs (26.1%) and the EMA an orphan designation to 20 (29.0%). Among these 69 novel drugs, 24 (34.8%) were first-in-class, 24 (34.8%) advance-in-class and 21 (30.4%) addition-to-class. The most prevalent therapeutic category was antineoplastic and immunomodulating agents (29% of all novel drugs from the sample) and many drugs (68.1%) were for chronic treatment. The manufacturer was a large pharmaceutical company for 44 (63.8%) of the drugs. Other details are in table 1.
Characteristics of 69 novel drugs approved by both the FDA and EMA between 2005 and 2010 (excluding everolimus and temsirolimus)
Number of postmarketing trials, status and patients recruited
Sequential exclusions leading to our final study sample of 6679 relevant postmarketing studies related to all 69 novel drugs are explained in a flow chart in online supplementary material S1. Characteristics of all postmarketing studies are shown in table 2. In all, 2901 studies (43.4%) were completed, 487 (7.3%) terminated, 1013 (15.2%) active yet not recruiting, 1895 (28.4%) recruiting and 319 (4.8%) not yet recruiting. When comparing respective numbers of postmarketing studies and all clinical studies (preapproval pivotal trials and postmarketing studies), the median proportion of postmarketing studies per drug was 0.96 (IQR 0.93–0.98). However, we found high variability in number of postmarketing studies per drug, with a median of 55 studies per drug (IQR 33–119) and mean of 96.8 studies per drug (SD 110.3). Galsulfase, an orphan medication indicated for mucopolysaccharidosis VI, was associated with the lowest number of postmarketing studies (n=3) and sorafenib, a tyrosine kinase inhibitor initially indicated for kidney cancer, with the highest number of postmarketing studies (n=530).
Characteristics of industry and non-industry postmarketing studies registered at ClinicalTrials.gov before 24 September 2014 for the 69 novel drugs in the study sample
Planned enrolment was also highly variable, with studies only including one patient, and one study intending to recruit 904 585 patients (actually a prospective population-based cohort study examining risk of congenital malformations after use of varenicline, a tobacco-use cessation drug, in pregnant women). However, the median number of patients to be enrolled per study was 60 (IQR 28–183). Data on the total population to be enrolled in all postmarketing studies for a given drug was also highly varied, with a median total sample of 15 418 patients (IQR 4932–37 523). Velaglucerase alfa, an orphan medication indicated for Gaucher disease, was associated with the lowest population size to be included in studies (n=67), and varenicline was associated with the greatest population to be enrolled (>1 million patients overall). Online supplementary material S2 shows the total number of patients to be included in postmarketing studies for each drug and proportions of industry and non-industry funders.
Supplementary file 3
Online supplementary material S3 presents for each drug the number of patients included in preapproval pivotal trials as compared with postmarketing studies. The median proportion for the population recruited in postmarketing studies to the total population (ie, preapproval samples and postmarketing studies) was 0.95 (IQR 0.90–0.98). Again, alglucosidase and velaglucerase alfa were associated with the lowest number of patients in preapproval pivotal trials. In contrast, for dabigatran, a drug initially indicated for preventing venous thromboembolism in the European Union and to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation in the USA, preapproval pivotal trials had recruited the highest number of patients. The same figure also shows the proportions of patients enrolled in postmarketing studies designed for the originally approved indication, another indication and both.
Supplementary file 4
Trial characteristics
Data regarding study phases are shown in table 2; only 18.6% of identified postmarketing studies were considered phase IV studies, whereas the most prevalent category was phase II studies (32.6%). Data regarding randomisation were missing for 2452 postmarketing studies (36.7%). Among the remaining studies for which these data were available, 3067 were randomised (72.6%). Other data are in table 2.
Sponsor
Industry funded or partially funded nearly two-thirds of postmarketing studies. Indeed, as shown in table 2, industry was the primary sponsor of 2713 studies (40.6%), but when also considering manufacturers as minority funders, industry was involved in a total of 4176 studies (62.5%). Data regarding postmarketing studies stratified by sponsorship are in table 2. Figure 1 presents the drug sample with respect to the number of postmarketing studies and the proportion of industry and non-industry funders for each drug. Online supplementary material S4 provides the same information but with a 4-year follow-up for each drug.
Supplementary file 5

Number of postmarketing studies and respective proportion of industry and non-industry funders.
Conditions addressed in trials
Review of indications showed that 2441 postmarketing studies (36.5%) were launched for another indication than the originally approved indication. Figure 2 displays the number of non-approved indications studied in postmarketing studies for each drug of our sample, with information regarding the more advanced phase for each newly targeted indication. When comparing those studies with the total number of clinical studies (preapproval pivotal trials and postmarketing studies), we found a median proportion of 0.24 (IQR 0.09–0.4). The median proportion for the population recruited in postmarketing studies designed for another indication than the originally approved indication to the total population from all clinical studies (preapproval pivotal trials and postmarketing studies) was 0.12 (IQR 0.03–0.33).

Number of non-approved indications targeted in postmarketing studies for each drug of our study sample. Indications are rank-ordered on the basis of the number of postmarketing studies launched (from the greatest number of postmarketing studies on the left side of the figure to the lowest number on the right side). Colour of boxes varies according to the advanced phase of the targeted indication. Indications are classified according to the Global Burden of Diseases classification.21 Indications belonging to residual categories or health conditions not relevant to the Global Burden of Diseases were excluded and therefore are not represented in the figure.
When analysing the relationship between the study sponsor and the study indication, we found that 68.7% of studies designed in the originally approved indication were sponsored by industry, as compared with 53.7% of studies designed in another indication (P<0.0001). Findings regarding planned enrolment according to the indication and stratified on funding origin are in online supplementary eTable. Regardless of the funder, postmarketing studies targeting originally approved indications planned to enrol more patients than those studying other indications.
Supplementary file 9
Timing
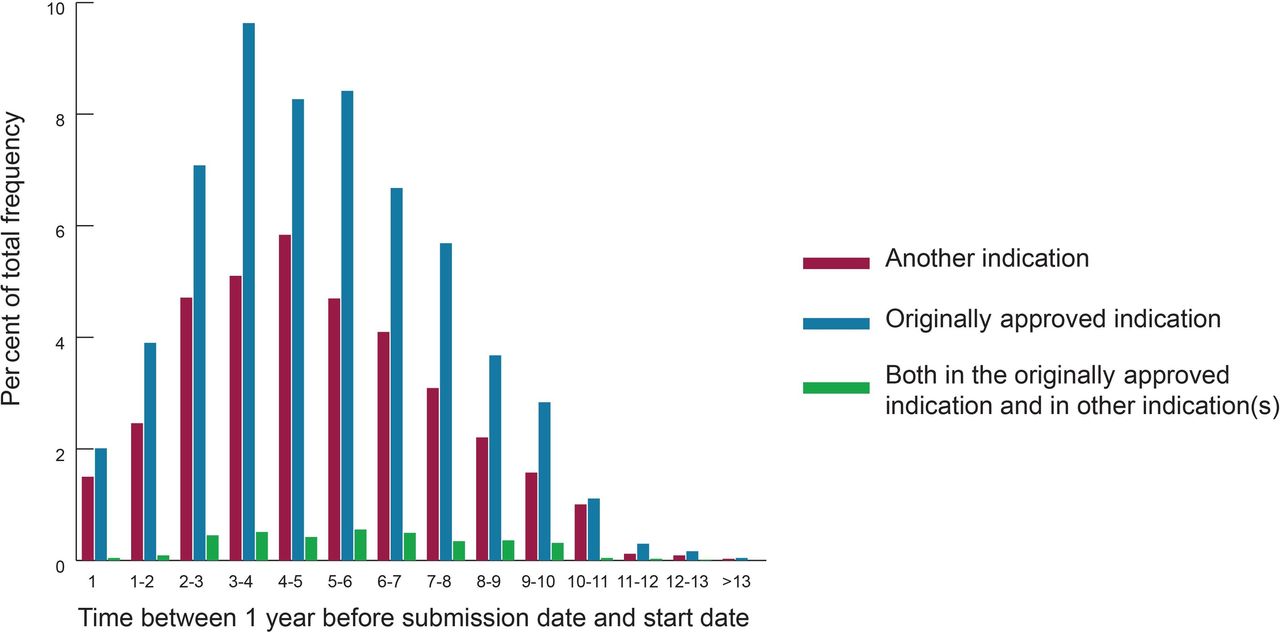
The annual number of postmarketing studies over the life-cycle of drugs, stratified by indication, is shown in figure 3, exhibiting an asymmetric bell pattern, with a rapid increase in number of postmarketing studies launched, a peak of activity within the third year after the first regulatory submission, then a progressive decline in number of launched studies. Detailed examination shows a greater proportion of studies designed for another indication than the originally approved indication at the beginning and end of drug life-cycle. Online supplementary material S5 is based on the same data but displays information regarding sponsors. Former postmarketing studies were predominantly funded by industry versus academic or not-for-profit entities and this proportion increased until the second year after the first regulatory submission. Afterwards, the proportion of non-industry funders tended to increase over time.
Supplementary file 6
![[SP3.jpg]](https://bmjopen.bmj.com/content/bmjopen/7/12/e018587/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
![[SP4.jpg]](https://bmjopen.bmj.com/content/bmjopen/7/12/e018587/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
![[SP5.jpg]](https://bmjopen.bmj.com/content/bmjopen/7/12/e018587/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
{kind=link}
{kind=link}
{kind=link}
Annual number of postmarketing studies over the life-cycle of drugs, stratified by indication.
Location
Overall, 80% of postmarketing studies were conducted in only one country. For 66 drugs, at least one study was conducted in at least two countries. Sorafenib was the most concerned drug in this regard, with 74 studies involving at least two countries. Data regarding locations of studies for each drug are in online supplementary material S6. In brief, postmarketing research was highly concentrated in North America (ie, USA and/or Canada; 44.8% of all postmarketing studies of the sample) and Europe (25.0%). Postmarketing studies conducted in other areas represented 15.6% of all studies, and studies conducted in multiple continents were few. When examining the relation between study location and study design with respect to the original label, we found that studies from North America (USA and/or Canada) were more frequently conducted for indications other than the originally approved indication versus those located in Europe (50.4% vs 36.9%). Data from online supplementary materials S2, S3 and S6 are summarised as online supplementary material S7 than the originally approved indication.
Supplementary file 7
![[SP7.jpg]](https://bmjopen.bmj.com/content/bmjopen/7/12/e018587/DC7/embed/inline-supplementary-material-7.jpg?download=true){kind=link}
Supplementary file 8
Supplemental indications
During the study period, 18 novel drugs (26.1%) were associated with a least a supplemental indication by the FDA: 1 with four supplemental indications, 1 with three supplemental indications, 5 with two supplemental indications and 11 with 1 supplemental indication. The mean time between the first regulatory submission and subsequent supplemental indication was 4.4 years (SD 1.7; IQR 3.3–5.7). The mean number of patients to be enrolled in postmarketing studies before approval of a supplemental indication was 12 763.1 (SD 12 474.3; IQR 3891.0–15 856.0).
Supplemental analysis
Analyses of postmarketing studies shown in table 2 were also performed when only taking into account those whose launch started after the first regulatory approval. Put another way, this supplemental set of analysis led us to exclude the 275 studies (see flow chart in online supplementary material S1) whose starting date had preceded the first regulatory submission by 1 year or less. Results are displayed in online supplementary material S8, showing no obvious difference with the main set of analysis.
Supplementary file 2
Discussion
In our study of postmarketing clinical research studies conducted for novel drugs approved by both the FDA and EMA between 2005 and 2010, we found high variability in number of postmarketing studies per drug and planned enrolment per study. Indeed, the median planned enrolment was low, 60 patients, with a median of 55 studies per drug, most of which had not yet been completed at a minimum of 4 years after approval. Locations were concentrated, with 72.3% of postmarketing studies conducted in North America and/or Europe and 80% conducted in only one country. Approximately 40% of postmarketing studies were designed for an indication other than the originally approved one, more frequently concerning studies not involving industry funding. Overall, those findings reflect the lack of global coordination of postmarketing research for novel drugs.
Our study has several strengths. First, we focused on a sample of drugs approved by the two leading medical product regulators, FDA and EMA, which suggests that these drugs are likely to be of the greatest interest and importance to clinicians worldwide. Most previous studies focused on the FDA or EMA but rarely both.20 22 Second, few comprehensive studies have analysed postmarketing research despite its undisputed public health impact.9–14 Most research focused on safety or was limited to a given therapeutic area, or even only one drug. In addition, we chose a large study period, with a 6-year span for drug approvals, and more than 10 years for the trial sample. Moreover, we followed a rigorous method for selecting postmarketing studies, excluding clinical trials included in the FDA submission, studies that had not been launched, studies mistakenly classified as involving the drug in ClinicalTrials.gov and studies whose starting date was too early as compared with regulatory submission. Third, we provide unique insights into the clinical research programmes examining non-approved drug uses. Many studies have investigated off-label prescriptions,23 24 but we used a slightly different approach. In effect, most drug labels are stringently phrased so as to be rigorously aligned to pivotal trial criteria.25 Therefore, categorising studies according to the actual off-label or on-label status of the drug investigated would have led to classifying most as involving off-label drug use. Put another way, the label was judged too narrow, and our method offers a more significant picture for clinicians and epidemiologists. We believe that our classification better reflects substantial evolution regarding the initially authorised use of novel drugs.
Our findings raise several issues worthy of consideration about postmarketing research. First, we showed that postmarketing research is both a heterogeneous and concentrated landscape, probably linked to its loose regulation6 and to market forces. Therefore, most initiatives are at the discretion of funders, either industry or academic institutions, and driven by various factors not necessarily linked to medical need or relevancy. For instance, prior research has shown that many postmarketing trials were ‘seeding trials’, designed for marketing purposes rather than scientific relevancy.7 8 The number of postmarketing studies per novel drug and planned enrolment were highly variable, but most studies were conducted in only one country and North America and Europe were by far the most frequent locations. Median planned enrolment was low and many studies were still not completed at the time of data acquisition. These findings question the absence of steering or the lack of effectiveness or incentive policies for postmarketing research. Second, almost 40% of postmarketing studies were designed for an indication other than the originally approved indication, with non-industry trials more likely concerned. Although industry has been blamed for testing their products in a too-liberal manner,26 our findings suggest that academics and other non-industry bodies might be more prone to assess authorised drugs in innovative ways to evaluate novel indications. Third, we found that postmarketing studies designed for the originally approved indication planned to enrol a greater number of patients on average than those targeting novel indications. This latter finding is somewhat reassuring because postmarketing studies for an already approved indication aim to refine knowledge regarding the long-term effect and/or safety and should therefore include more patients than preapproval pivotal trials.
Our study has limitations. The first may be a registration bias at ClinicalTrials.gov, which would alter the exhaustiveness of our assessment. Some studies are not registered by researchers27 28 and were therefore not included in our study. Others are imperfectly registered, with some information missing. However, ClinicalTrials.gov is widely recognised as a benchmark registry, and recent reports showed that compliance might have improved over time.29 Another limitation is the definition of postmarketing studies, in that clinical studies are designed and launched according to a continuous timing and a single threshold might be lacking for distinguishing premarketing and postmarketing trials. Therefore, we decided to consider studies starting at most 1 year before the first regulatory submission as postmarketing studies even though we could have made another choice. A third limitation is related to data sources. For some data, we relied on only one of the two selected regulators. We used such an approach for the sake of convenience and recognise that this could be interpreted as a bias, yet to our knowledge, there are very few if any differences in data between the two studied regulators. Therefore, this latter limitation in the methods seems unlikely to affect our findings. Finally, we could not identify whether postmarketing trials were relevant or useful because we did not analyse their design, endpoints or comparators, among other factors.
In conclusion, our research shows that postmarketing research is highly variable and concentrated, with on one hand, great differences in the number of postmarketing studies per drug and in planned enrolment and on the other, most studies being conducted in only one country, with North America and Europe the most represented locations. Approximately 40% of postmarketing studies assessed the drug for an indication other than the originally approved indication, more frequently non-industry studies. Even though some of our findings can be seen as reassuring, others underline the lack of global coordination of postmarketing research for novel drugs despite the undisputed influence of such research.
Acknowledgments
The authors are grateful to EMA officials who graciously helped them classify drug manufacturers. They also thank Geoffroy Beraud-Chaulet for his useful work on Drugs@FDA and Elise Diard for her invaluable help on figures.
References
Footnotes
Contributors J-DZ and PR were responsible for the conception and design of this work. J-DZ drafted the manuscript and was responsible for most of the data acquisition. IA was responsible for data exportation and structuration. AV was responsible for collection of some data and contributed to categorisation of studies. NSD was responsible for some of the data acquisition. GB conducted the statistical analysis. JSR and PR provided supervision. All authors participated in the analysis and interpretation of the data and critically revised the manuscript for important intellectual content.
Competing interests J-DZ reports that he serves as an advisor for several consulting firms and communication companies linked with the pharmaceutical industry (Cepton, Oliver Wyman, Roland Berger, McCann Healthcare, Omnicom, Grey Healthcare, Saatchi and Saatchi Healthcare, Sudler & Hennessey, TBWA, inVentiv Health France, Havas). He also reports compensation for lectures given to manufacturer professional associations; collaboration with Mayoly-Spindler, Merck, Teva, Johnson & Johnson and Menarini; unpaid consultancy for EY; conducting workshops funded by Amgen; and being invited to a French medical congress by AbbVie. JSR receives support through Yale University from Johnson & Johnson to develop methods of clinical trial data sharing; from the Centers of Medicare and Medicaid Services (CMS) to develop and maintain performance measures that are used for public reporting; from Medtronic and the US FDA to develop methods for postmarket surveillance of medical devices; from the Blue Cross Blue Shield Association to better understand medical technology evaluation and from the Laura and John Arnold Foundation to support the Collaboration on Research Integrity and Transparency (CRIT) at Yale.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data files are available from the corresponding author on reasonable request.