Article Text

Abstract
Introduction Infants born late preterm (34+0 to 36+6 weeks’ gestational age) have frequent episodes of intermittent hypoxaemia compared with term infants. Caffeine citrate reduces apnoea and intermittent hypoxaemia and improves long-term neurodevelopmental outcomes in infants born very preterm and may have similar effects in late preterm infants. Clearance of caffeine citrate increases with gestational age and late preterm infants are likely to need a higher dose than very preterm infants. Our aim is to determine the most effective and best-tolerated dose of caffeine citrate to reduce transient intermittent hypoxaemia events in late preterm infants.
Methods and analysis A phase IIB, double-blind, five-arm, parallel, randomised controlled trial to compare the effect of four doses of oral caffeine citrate versus placebo on the frequency of intermittent hypoxaemia. Late preterm infants will be enrolled within 72 hours of birth and randomised to receive 5, 10, 15 or 20 mg/kg/day caffeine citrate or matching placebo daily until term corrected age. The frequency of intermittent hypoxaemia (events/hour where oxygen saturation concentration is ≥10% below baseline for ≤2 min) will be assessed with overnight oximetry at baseline, 2 weeks after randomisation (primary outcome) and at term corrected age. Growth will be measured at these timepoints, and effects on feeding and sleeping will be assessed by parental report. Data will be analysed using generalised linear mixed models.
Ethics and dissemination This trial has been approved by the Health and Disability Ethics Committees of New Zealand (reference 18/NTA/129) and the local institutional research review committees. Findings will be disseminated to peer-reviewed journals to clinicians and researchers at local and international conferences and to the public. The findings of the trial will inform the design of a large multicentre trial of prophylactic caffeine in late preterm infants, by indicating the most appropriate dose to use and providing information on feasibility.
Trial registration number ACTRN12618001745235; Pre-results.
- neonatology
- clinical pharmacology
- paediatric thoracic medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This study seeks to address the rates of neurodevelopmental impairment among late preterm infants by investigating the optimal dose of caffeine, as a potential primary neuroprotective strategy.
This is the first randomised placebo-controlled trial of four different doses of caffeine for the prevention of intermittent hypoxaemia in late preterm infants.
A strength of the trial is that both clinicians and parents will be blinded to treatment allocation with all groups receiving the same volume of an identical-appearing masked suspension.
The success of the trial depends on high compliance of administration of study medication by caregivers, which will be monitored by intermittent measurement of study bottle contents and infant salivary caffeine concentrations.
Future studies will be required to determine if caffeine reduces neurodevelopmental impairment in late preterm infants, based on the optimal dose determined by this trial.
Introduction
Late preterm infants are those born between 34 weeks and 36 weeks and 6 days’ gestation.1 They form the largest group within the preterm population, accounting for 68% of all preterm births or 5% of all births in New Zealand2 and 7% of all births in the USA.3 Late preterm infants are physiologically and metabolically immature1 and have a higher risk of morbidity and mortality in the neonatal period than full-term infants.4 They are more likely than full-term infants to have delayed establishment of oral feeding, temperature instability, hypoglycaemia, jaundice and respiratory distress and to undergo investigation for sepsis.5 Despite these risks, their size and weight mean they are often managed in a similar manner to full-term infants and cared for on postnatal wards rather than in neonatal units6 and are not treated with the routine prophylactic interventions such as caffeine, nutritional supplements and probiotics that are common practice in very and extremely preterm infants.
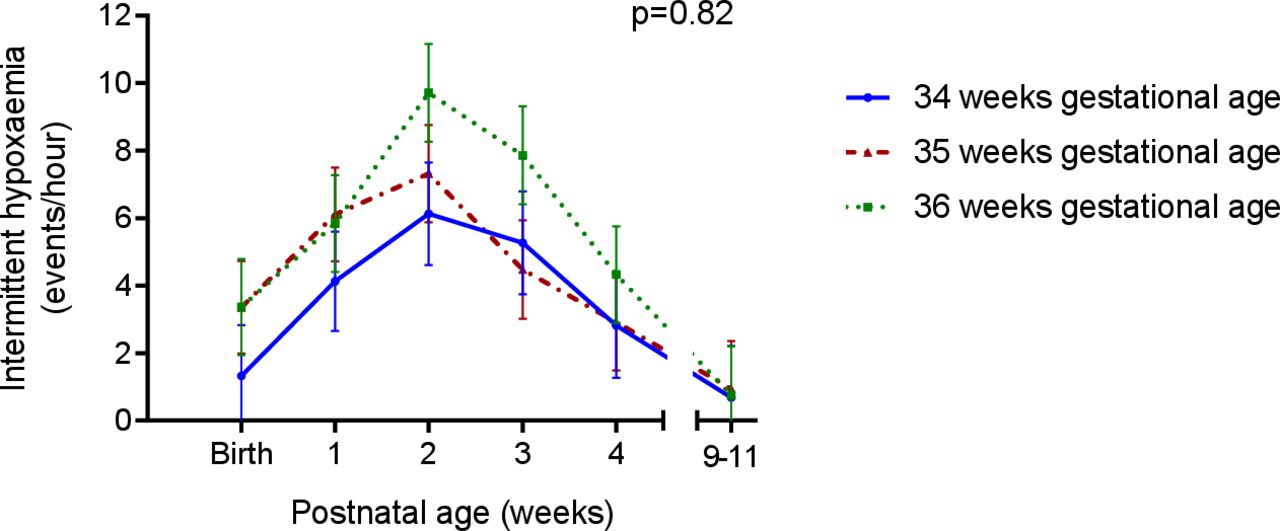
Apnoea of prematurity are prolonged pauses in breathing, of 20 s or more, which may cause a reduction in the oxygen saturation and bradycardia, and are associated with an increased incidence of brain injury7 and neurodevelopmental impairment.8 Late preterm infants experience apnoea of prematurity, although less frequently than in very preterm infants.9 Recently, we have demonstrated that late preterm infants have frequent episodes of intermittent hypoxaemia,10 transient repetitive decreases in oxygen saturation not associated with apnoea, but potentially causing similar organ hypoxia. In late preterm infants, the frequency of intermittent hypoxaemia peaks at 2 weeks’ postnatal age, before reducing to baseline levels at term corrected age (figure 1).10

Rate of intermittent hypoxaemia in late preterm infants in the 9–11 weeks following birth; adapted from Williams et al.10
Studies in adults have consistently shown that even brief exposure to hypoxia, whether from high altitude11 or carbon monoxide poisoning,12 can have long-term adverse effects on cognition. Even small changes in oxygen saturations in the neonatal period have been shown to significantly affect survival and neurodevelopment of very preterm infants.13–15 Intermittent hypoxaemia is associated with altered brain development in neonatal mice16 and reduced cognition and behavioural deficits in neonatal rats.17 In humans, transient intermittent hypoxaemic events are associated with poor neurodevelopmental outcomes in extremely preterm infants18 and in children with sleep-disordered breathing19 and congenital heart disease.20
Caffeine is a respiratory stimulant that is effective in the prevention and treatment of apnoea of prematurity and intermittent hypoxaemia and also reduces the incidence of chronic lung disease, cerebral palsy and cognitive delay in very preterm infants.21–23 Follow-up to 11 years of age has recently shown that caffeine treatment reduces the risk of motor dysfunction by a third infants born very preterm.24 25 While some of the long-term beneficial effects of caffeine may be due to its effect in reducing the incidence of bronchopulmonary dysplasia,26 there is also benefit from reducing the amount of time that infants are hypoxic, independent of the effect on bronchopulmonary dysplasia.27 Thus, caffeine has become the standard of care for very preterm infants and is in widespread use in neonatal units around the world as one of the few neonatal treatments that has been proven to have long-term neurodevelopmental benefit and also to be very well tolerated.
In the longer term, late preterm infants are more likely to be diagnosed with cerebral palsy,28 29 developmental delay,30 31 cognitive impairment32–34 and behavioural disorders35 compared with their term-born peers. However, few studies have investigated interventions to improve the neurodevelopmental outcomes of late preterm infants. As late preterm infants have an increase in hypoxaemic events compared with term infants, hypoxaemic events are associated with poor neurodevelopmental outcomes, it is possible that caffeine, an intervention that reduces hypoxaemic events and has already been shown to improve long-term outcomes in extremely and very preterm infants, may be effective at improving outcomes in late preterm infants.
In adults, most caffeine metabolism is via cytochrome P450 1A2 in the liver.36 However, in newborn preterm infants, hepatic metabolism of caffeine is almost absent, and most caffeine is eliminated via the kidneys, which are also immature. Therefore, caffeine elimination is slow in extremely preterm infants, and the half-life of caffeine is long. With increasing postconceptial age, the elimination of caffeine increases,37 38 and larger doses may be needed to maintain a therapeutic effect. However, the pharmacokinetic studies of caffeine in preterm infants to date have been done to treat apnoea in very preterm infants, rather than to treat intermittent hypoxaemia in late preterm infants.39
There is a wide range in the dose of caffeine citrate given to extremely preterm infants, from daily doses of 5 mg/kg22 to 20 mg/kg.40 The Caffeine for Apnoea of Prematurity trial used a dose of 5 mg/kg, which could be increased to 10 mg/kg if necessary to control apnoea of prematurity.22 The trial by Rhein et al found that in very preterm infants, 6 mg/kg of caffeine citrate reduced intermittent hypoxaemia at 35 weeks’ and 36 weeks’ postmenstrual age, but not after 36 weeks’ postmenstrual age.41 The authors hypothesised that this may have been due to an insufficient dose as the infants matured. Therefore, the most effective dose of caffeine to treat intermittent hypoxaemia in late preterm infants is unknown.
In very preterm infants, caffeine is usually well tolerated, but occasionally infants on caffeine develop tachycardia and feed intolerance.40 Caffeine also causes reduced neonatal weight gain compared with placebo,22 and in ventilated preterm infants, a higher dose of caffeine citrate (20 mg/kg) leads to reduced weight gain compared with a low dose (5 mg/kg).40 As in adults, infants on caffeine can develop irritability, sleeplessness and gastrointestinal disturbance. For caffeine to be used as a prophylactic medication in a large number of late preterm infants, it will need to be prescribed at a dose that has a low risk of significant side effects.
We are therefore undertaking the Latte Dosage Trial, a randomised, placebo-controlled dosage trial, to determine the most effective and best-tolerated dose of oral caffeine citrate to reduce intermittent hypoxaemia in late preterm infants.
Aim
To determine the most effective and best-tolerated dose of caffeine citrate to reduce intermittent hypoxaemia in late preterm infants.
Hypothesis
Caffeine citrate will reduce the frequency of intermittent hypoxaemia in late preterm infants in a dose-dependant manner.
Methods and analysis
Study design
The Latte Dosage Trial is a phase IIB, double-blind, five-arm, parallel, randomised controlled trial to compare the effect of four different doses of oral caffeine citrate versus placebo on the frequency of intermittent hypoxaemia in late preterm infants.
Recruitment and randomisation
Participants will be recruited by trial investigators or study nurses/midwives within 72 hours of birth from the neonatal unit and postnatal wards at Auckland City and Middlemore Hospitals in Auckland, New Zealand. Following written informed consent and enrolment, trial participation may occur in hospital, at a primary maternity unit or at home, as the patient’s clinical care dictates. Eligible participants are those infants born between 34 weeks and 36 weeks’ and 6 days gestation without contradiction to caffeine treatment, with the following exclusions:
Major congenital abnormality.
Minor congenital abnormality likely to affect respiration, growth or development.
Previous caffeine treatment.
Renal or hepatic impairment.
Tachyarrhythmia.
Seizures.
Hypoxic ischaemic encephalopathy.
Residing outside of the Auckland region.
Infants will be assigned randomly via an internet randomisation service (Clinical Data Research Hub, Liggins Institute, University of Auckland) to receive either daily caffeine citrate 5 mg/kg, 10 mg/kg, 15 mg/kg, 20 mg/kg or placebo. The allocation sequence will be generated by the study statistician, with 1:1:1:1:1 allocation stratified by study site and gestational age at birth (34, 35 or 36 weeks) (figure 2) using variable block sizes, with infants from multiple births being randomised to the same treatment group.

{kind=link}
{kind=link}
Flow diagram of randomisation schedule.
Different concentrations of the caffeine citrate (5 mg/mL, 10 mg/mL, 15 mg/mL and 20 mg/mL) will be prepared in identical bottles to the placebo so that all infants receive the same dose volume (2 mL/kg loading dose followed by 1 mL/kg, once a day). Bottles will be labelled in randomisation blocks using a lettering system that will change halfway through the study in order to maintain concealment from study personnel.
Study intervention
The infant will be given an enteral loading dose of the study drug (10 mg/kg, 20 mg/kg, 30 mg/kg or 40 mg/kg of caffeine citrate or water) in the morning after baseline oximetry (ie, prior to the infant reaching 96 hours of age), followed by a daily dose each morning (5 mg/kg, 10 mg/kg, 15 mg/kg or 20 mg/kg of caffeine citrate or placebo) until term equivalent age (40 weeks’ postmenstrual age). The dose will be recalculated weekly for weight after the infant has regained birth weight using the weight recorded by study staff at 2 weeks after randomisation and those recorded by usual care providers between 2 weeks’ and term corrected age. The study drug will be given via a nasogastric tube for infants with a tube in situ and orally for infants who do not require a nasogastric tube. Infants who are not able to tolerate enteral medications will have the study drug withheld until they are able to tolerate enteral intake.
Compliance will be assessed by measurement of study medication remaining in the bottle. At the 2-week visit, study staff will collect the initial bottle(s) issued at the start of the study and replace it with a new bottle(s), which will in turn be collected at the final visit. Liquid remaining in the bottle on each occasion will be measured and compared with the expected volume to assess compliance. Good compliance will be defined as ≥80% of the expected volume having been removed from the bottle. At the final visit, parents will be asked which treatment they think their infant received to assess the adequacy of blinding.
Apart from the study intervention and associated assessments, all other clinical cares, including the decision on when to discharge participants from the hospital and/or primary birthing units, will continue to be provided by the local clinical team, in accordance with usual guidelines and practices. Should an infant participating in the study require treatment for apnoea or intermittent hypoxaemia, clinicians will be encouraged to use oxygen or positive pressure ventilation as first-line treatments. If necessary, a loading dose of caffeine citrate may be given. If ongoing treatment with caffeine is necessary for the opinion of the treating clinician, they can discuss the option of partially unblinding the infant (caffeine or placebo) with the site principal investigator. If unblinding is required, information on the allocation of the participant will be communicated by the data management team to the treating clinician or pharmacist (who may inform the parents if requested), with the research team remaining blinded. Clinical open-label use of caffeine will be recorded. Unblinded infants will remain in the study unless parents request withdrawal, with infants analysed on an intention-to-treat basis.
Outcomes
The primary outcome for this study is the frequency of intermittent hypoxaemia (events/hour, defined as a brief transient fall in oxygen saturation concentration ≥10% below baseline) on overnight oximetry, 2 weeks after randomisation. Events longer than 2 min are considered a change in baseline rather than a transient desaturation event. Transient intermittent hypoxaemic events, if frequent or severe, are thought to have neurocognitive effects as significant as prolonged hypoxaemia.20 42 Although a 3% threshold is used in polysomnography to define desaturation events, a definition of 10% is commonly used in the neonatal literature. In addition, due to the variability of events, we considered a 10% threshold more repeatable and reliable than a 3% threshold for defining events.
Secondary outcomes include:
Respiratory: frequency of intermittent hypoxaemia on overnight oximetry at term equivalent age; mean overnight oxygen saturation at 2 weeks and term equivalent age; use of respiratory support, including oxygen, until term equivalent age.
Growth: growth velocity from birth to term equivalent age for weight gain, length and head circumference; failure to regain birth weight by 2 weeks of age.
Side effects: feed intolerance as reported by parents;43 duration of tube feeding; sleep and arousal as reported by parents (measured by subscale nine on the Infant Behaviour Questionnaire-Revised, modified for neonates44); tachycardia; study drug stopped due to presumed side effects; neonatal seizures requiring anticonvulsant treatment before 44 weeks postmenstrual age; neonatal or infant death.
Maternal and infant salivary caffeine concentration at 2 weeks after randomisation.45
Readmission to hospital until 44 weeks postmenstrual age or open-label caffeine use.
Maternal caffeine intake at birth, 2 weeks and term corrected age and mental health (Edinburgh postnatal depression score) at birth and term corrected age.46
The timing of the study intervention and assessments is summarised in table 1.
Study intervention and assessment
Data collection methods
Online data management services will be provided by the Clinical Data Research Hub (Liggins Institute, University of Auckland). Data collection will use the REDCap platform (Vanderbilt University) for clinical report forms, with password-protected secure servers used to store data.
Pulse oximetry
Overnight pulse oximetry (Rad 8, Masimo, Irvine, California, USA) will be recorded for a period of 12 hours from either foot at baseline, 2 weeks after randomisation (range 12–21 days) and at term corrected age (range 40–41 weeks postmenstrual age) using a 2 s averaging time and 2 s resolution. Recordings will be conducted at home, in primary birthing units or at home as dictated by the clinical care requirements of the participants. Where recordings are conducted at home, parents will be visited the day that recording starts by a member of the research team. The oximeter will be set up, and the parents will receive instruction in attaching the probe to the baby’s foot and be instructed to do this when placing the baby down to sleep in the evening. If necessary, the research team member may visit late in the day to apply the probe or provide support via a video call to ensure this is done correctly by parents. Unless clinically required, oximeters will be operated in sleep mode, with no displays or alarms. The oximetry recording will be downloaded with PROFOX oximetry software (V.Masimo 2011.27D, PROFOX Associates, Esconditso, California, USA) and edited to remove readings with poor signal or aberrant data. Only recordings with more than 6 hours will be included in the analysis, recordings with less than 6 hours of edited data will be repeated the following night.
Anthropometry
Weight, length and head circumference will be measured at study entry and at the 2-week and term visits, with birth weight and neonatal centiles calculated using Fenton-WHO growth charts for preterm infants,47 and growth velocity will be calculated between birth and term equivalent using an exponential model.48
Salivary caffeine concentrations
Two weeks after randomisation, a saliva sample will be collected from infants for assessment of caffeine concentration. Samples will be taken using a mouth swab prior to administration of the morning dose of trial medication. In 24 hours preceding this, mothers will be asked to collect three saliva samples by spitting into collection tubes at three predetermined time points during the day, with the mean of these three samples used to determine average daytime maternal salivary caffeine concentration. Collection of these samples will allow us to compare maternal and infant salivary caffeine concentrations to establish whether maternal caffeine intake contributes significantly to infant caffeine levels via breastfeeding or not and to help assess compliance with the study intervention.
Questionnaires
Mothers will complete questionnaires to provide demographic and contact details at enrolment and to assess smoking, infant feeding and sleeping, maternal caffeine intake and maternal mental health as detailed in table 1.
Neonatal morbidity
Information on neonatal morbidity, including supplemental oxygen, respiratory support and apnoea requiring stimulation, will be recorded from the neonatal record. Exposure to antenatal corticosteroids will be recorded.
Discontinuation of intervention/ withdrawal
The allocated treatment may be stopped at any time by the parents or the clinician caring for the infant if they feel that this is in the best interests of the infant, without formally withdrawing, in which case data collection will continue and results will be analysed on an intention-to-treat basis.
Should a parent wish to withdraw from the study, they will have the option of:
Discontinuation of study drug, with continuation of collection of minimum outcome data.
Withdrawal from the study and discontinuation of further data collection, with data collected prior to withdrawal used.
Complete withdrawal from the study, with removal of previously collected data.
Patient and public involvement
The Latte Dosage Study methodology was discussed, developed and refined as part of the 2017 On-Track Network clinical trial development workshop that included consumer and Māori cultural advisor input. Perinatal consumer representatives provided advice and input into the development of the clinical trial protocol.
Sample size
Based on our previous study,10 we estimate a background mean (SD) frequency of 6.9 (3.4) episodes of intermittent hypoxaemia per hour at 2 weeks’ postrandomisation. To detect a 50% reduction of 3.5 episodes per hour with 90% power, allowing for a 10% drop out rate and clustering of multiples with an intraclass correlation coefficient of 0.05, we will require 24 infants in each of the five arms (total 120 infants), with two-sided alpha of 0.05. Recruitment to the study started in February 2019 and is scheduled to conclude in December 2020.
Data analysis
The primary analysis will compare primary and secondary outcomes between groups using generalised linear mixed models with adjustment for gestational age at birth and site (fixed effects), non-independence of multiples (random effect) and pairwise comparisons between the different caffeine groups and between the caffeine groups and the placebo group using Dunnett’s multiple comparison test. The selection of the optimal dose will be based on a combination of the dose with the greatest reduction of intermittent hypoxaemia with a minimum number of side effects and a pragmatic consideration of the ease of administration. Linear trends, such as growth, will be tested using orthogonal contrasts. In keeping with Consolidated Standards of Reporting Trial recommendations,49 baseline imbalance between babies in the randomised groups will not be formally tested. Edinburgh Postnatal Depression Scale scores will be adjusted for baseline values. Categorical data will be presented as number and per cent, and continuous data as mean and SD or median and IQR, as appropriate. Denominators will be given for all outcomes. Treatment effects will be presented as OR, count ratio, mean difference or ratio of geometric means (positively skewed data), as appropriate, with 95% CIs. All tests will be two tailed, with p<0.05 considered significant. The data will be analysed on an intention-to-treat basis.
The following secondary analyses will be performed:
Compliance: a per-protocol analysis will be performed for the primary outcome that includes only those infants who were compliant with the study drug.
Open-label caffeine treatment: a sensitivity analysis will be performed for the primary outcome that includes only those infants who did not receive additional open-label caffeine treatment.
Maternal caffeine: an exploratory analysis will be performed on the effect of maternal caffeine intake on the primary outcome by performing additional adjustments for maternal caffeine intake from the questionnaire and maternal salivary caffeine concentration. For infants who are fully formula fed, infant caffeine exposure to maternal caffeine intake will be assumed to be zero.
An independent data monitoring committee will review trial data after enrolment of 60 infants to the trial. The data monitoring committee provides advice to the trial steering group on any modifications that may be required. There are no formal stopping guidelines.
Ethics and dissemination
Ethical approval has been obtained from the Health and Disability Ethics Committees of New Zealand (reference 18/NTA/129) and by the local institutional research review committees for each centre. The trial is registered with the Australian New Zealand Clinical Trials Registry from 24 October 2018.
The results of the trial will be published in an international peer-reviewed journal and disseminated via presentations at local and international conferences to researchers and clinicians. A lay summary of the research findings will be made available to those parents who indicated a wish to receive these on their consent forms.
Discussion
Late preterm infants experience higher rates of intermittent hypoxaemia than their term-born peers and have poorer long-term neurodevelopmental outcomes.28–35 Caffeine is well established as a treatment for apnoea of prematurity in very and extremely preterm infants and improves long-term neurodevelopmental outcomes in these infants.21 50 51 Caffeine use in late preterm infants may also reduce episodes of intermittent hypoxaemia and improve long-term outcomes for these infants. As late preterm infants make up the majority of preterm infants, interventions that improve long-term outcomes in this population are likely to have the greatest public health impact in terms of interventions for preterm infants.52
The Latte Dosage Trial seeks to establish the most effective and best-tolerated dose of caffeine citrate for the prevention of intermittent hypoxaemia in late preterm infants. It is the first trial to investigate the use of caffeine, an inexpensive medication already widely used in neonatal care, for this indication. Data from the Latte Dosage Trial will be used to inform the development of a large-scale, multicentre trial investigating the efficacy of caffeine treatment in late preterm infants in preventing neurodevelopmental impairment by indicating the most appropriate dose to use and providing information on feasibility.
Acknowledgments
Brian Darlow and Members of the On-Track network for assistance in developing and refining the study design. Sarah Phillipsen, Sabine Huth, Lisa Mravicich and Florella Keen for assistance in setting up the trial. Sara Hanning and Trusha Purohit for advice on caffeine testing and development of the caffeine assay. Alana Cavadino for statistical advice.
References
Footnotes
Contributors JMA conceived and developed the study design, drafted the original study protocol, approved the final study protocol and reviewed the article for publication. EAO contributed to the study design, approved the final version of the study protocol and drafted the article for publication. CJDM contributed to the study design, approved the final version of the study protocol and reviewed the article for publication. DGM contributed to the study design, approved the final version of the study protocol and reviewed the article for publication.
Funding This work is supported by the Health Research Council of New Zealand: grant number 18/613.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.